溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這期內容當中小編將會給大家帶來有關怎么使用MutationalPatterns進行腫瘤突變頻譜分析,文章內容豐富且以專業的角度為大家分析和敘述,閱讀完這篇文章希望大家可以有所收獲。
MutationalPatterns是一個bioconductor上的R包,可以用于腫瘤突變頻譜的分析。腫瘤突變頻譜針對點突變進行定義,A,T,C,G四種堿基兩兩突變,共有4X3=12種排列,考慮到正負鏈堿基配對原則,正鏈上的A->C突變,對應負鏈上為T->G, 所以進一步轉換成了一個組合的問題,所以某個位點的突變可以劃分為以下6種模式
C>A, 表示C>A和G>T兩種
C>G, 表示C>G和G>C兩種
C>T, 表示C>T和G>A兩種
T>A,表示T>A和A>T兩種
T>C,表示T>C和A>G兩種
T>G,表示T>G和A>C兩種
進一步考慮突變位點所處的序列上下文環境,即上下游各取一個堿基再加上突變位點的堿基,組成了3個堿基的motif, 可以有4X4X6=96種模式,每種模式的頻率分布就是突變頻譜。突變頻譜可以當做一個腫瘤樣本的特征,進行樣本間的比較。通過MutationalPatterns包,可以方便的根據樣本對應的VCF文件,提取突變頻譜的信息,首先讀取文件,代碼如下
# 加載R包
> library(MutationalPatterns)
# 列出vcf的路徑
> vcf_files <- c("sample1.vcf", "sample2.vcf")
# 設置vcf文件對應的樣本名稱
> sample_names <- c("sample1", "sample2")
# 加載參考基因組
> library(BSgenome.Hsapiens.UCSC.hg19)
> ref_genome <- "BSgenome.Hsapiens.UCSC.hg19"
# 讀取vcf文件
> vcfs <- read_vcfs_as_granges(vcf_files, sample_names, ref_genome)讀取完成之后,可以先統計下6種不同的點突變模式的分布,代碼如下
> type_occurrences <- mut_type_occurrences(vcfs, ref_genome)
> plot_spectrum(type_occurrences)可視化結果示意如下
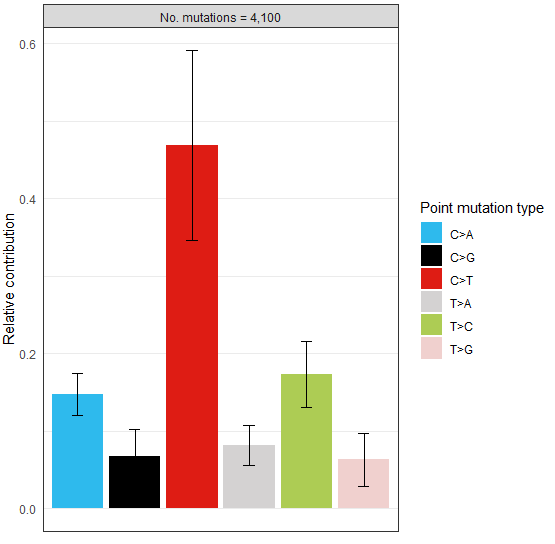
該R包經典的使用場景如下
根據vcf文件,計算每個樣本中96種motif的頻數,并可視化,代碼如下
> mut_mat <- mut_matrix(vcf_list = vcfs, ref_genome = ref_genome)
> plot_96_profile(mut_mat[,c(1,2)], condensed = TRUE)可視化結果示意如下
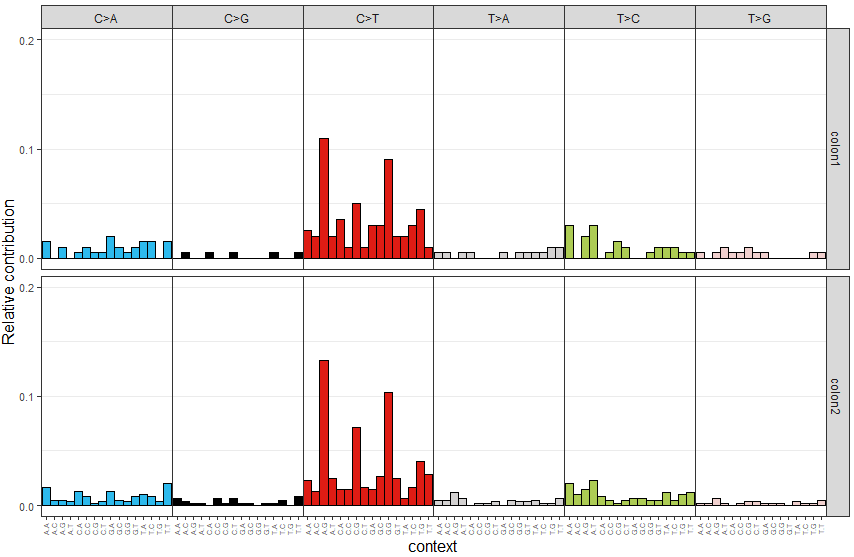
代碼如下
> plot_compare_profiles(mut_mat[,1], mut_mat[,2], condensed = TRUE)
可視化結果示意如下
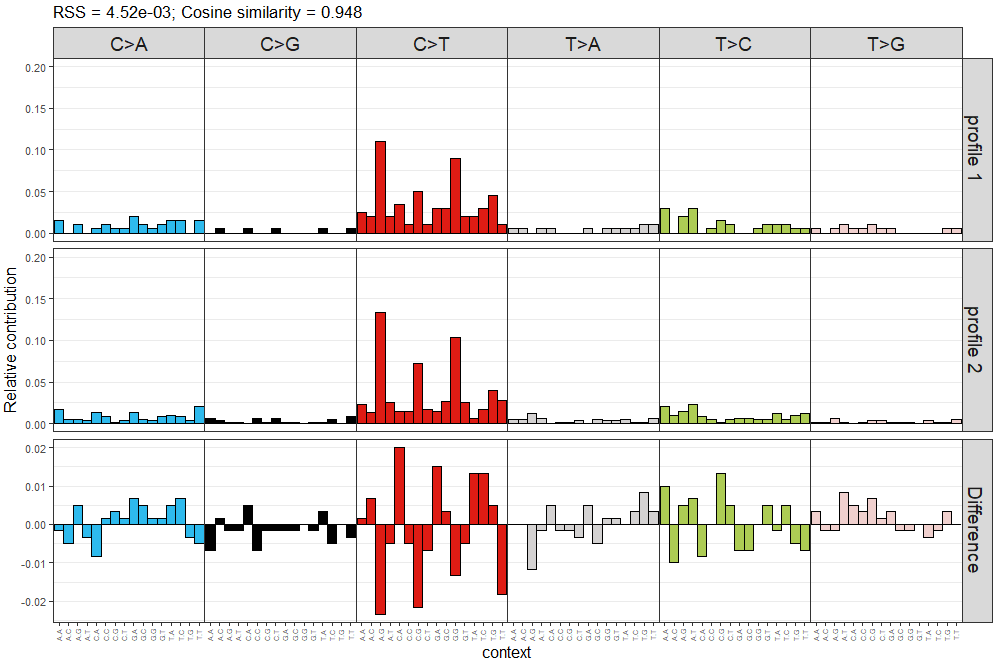
左上角的給出了兩個頻譜間cosine similarity相似度,圖片中前兩層分別對應兩個需要比較的頻譜,第三層為兩個頻譜的差異,直接用頻率相減。
通過非負矩陣分解NMF算法,從原始的突變頻譜中提取特征,稱之為突變特征mutation signature,代碼如下
> library(NMF)
> estimate <- nmf(mut_mat, rank=2:5, method="brunet", nrun=10, seed=123456)
> nmf_res <- extract_signatures(mut_mat, rank = 2, nrun = 10)
> colnames(nmf_res$signatures) <- c("Signature A", "Signature B")
> rownames(nmf_res$contribution) <- c("Signature A", "Signature B")
> plot_96_profile(nmf_res$signatures, condensed = TRUE)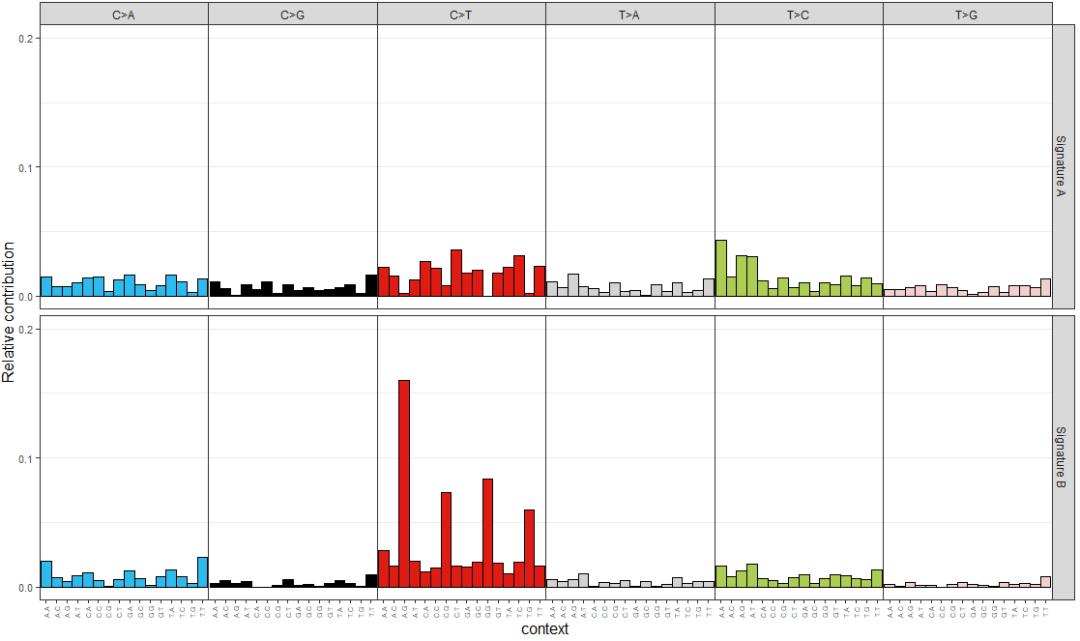
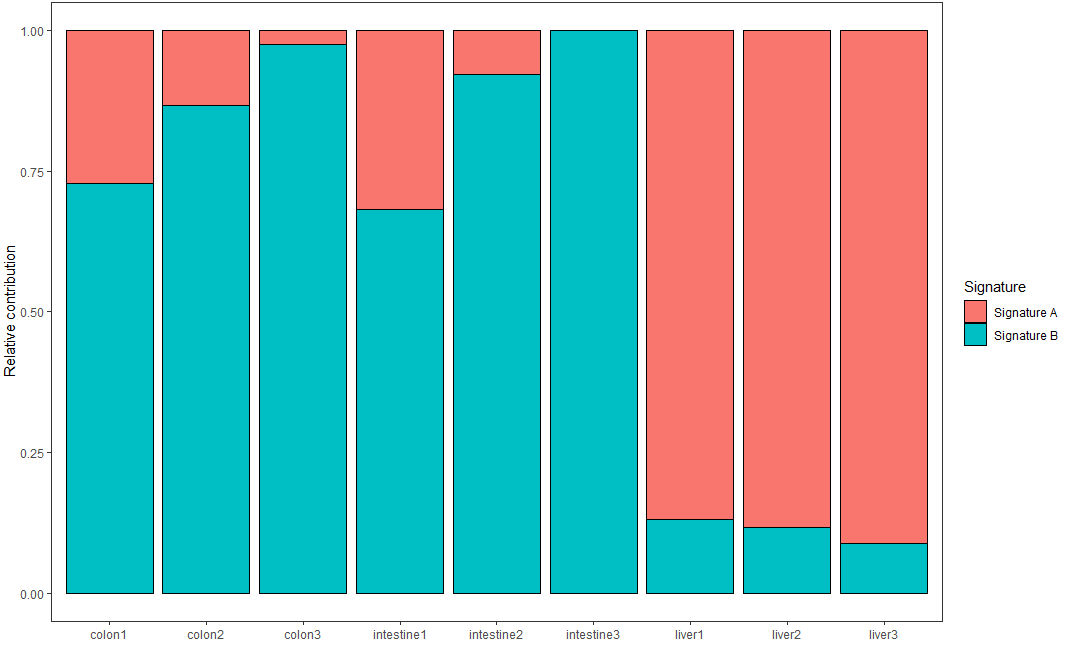
每個樣本的突變頻譜是不同突變特征組成的結果,通過如下代碼可視化每個樣本中不同突變特征的貢獻率
plot_contribution(nmf_res$contribution, nmf_res$signature, mode = "relative")
可視化結果示意如下
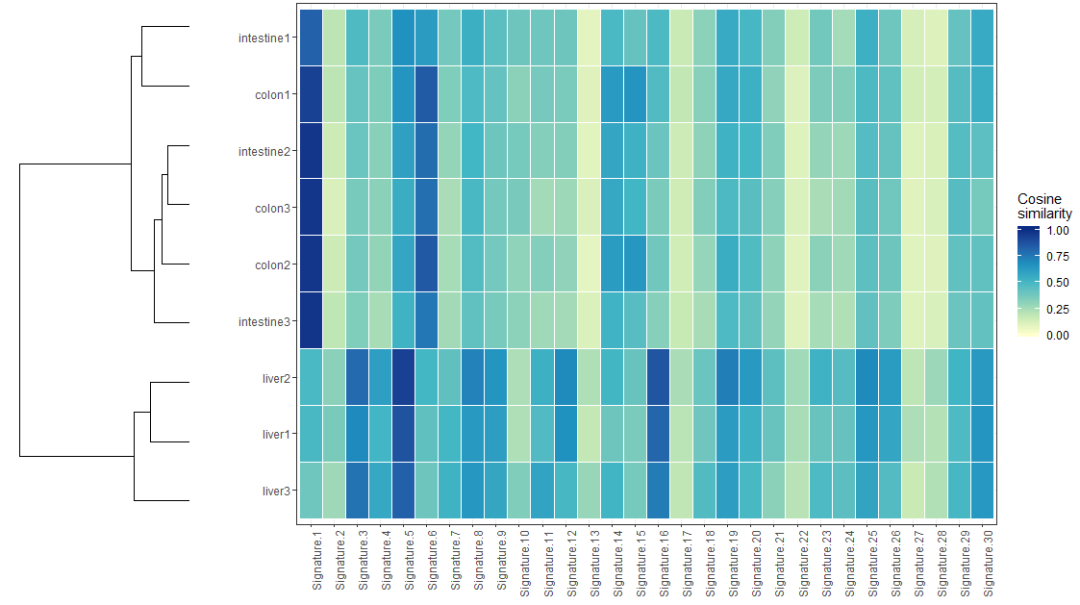
計算圖片頻譜間的cosine similarity相似度,結果用熱圖展現,代碼如下
> cos_sim_samples_signatures = cos_sim_matrix(mut_mat, mut_mat)
> plot_cosine_heatmap(cos_sim_samples_signatures)可視化的結果示意如下
通過這個R包,可以輕松實現突變頻譜的常見分析內容。
上述就是小編為大家分享的怎么使用MutationalPatterns進行腫瘤突變頻譜分析了,如果剛好有類似的疑惑,不妨參照上述分析進行理解。如果想知道更多相關知識,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





