溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要講解了“如何使用ChIPseeker進行peak注釋”,文中的講解內容簡單清晰,易于學習與理解,下面請大家跟著小編的思路慢慢深入,一起來研究和學習“如何使用ChIPseeker進行peak注釋”吧!
ChIPseeker是使用的最廣泛的peak注釋軟件之一,提供了以下多種功能
peak在染色體和TSS位點附近分布情況可視化
peak關聯基因注釋以及在基因組各種元件上的分布
獲取GEO數據庫中peak的bed文件
多個peak文件的比較和overlap分析
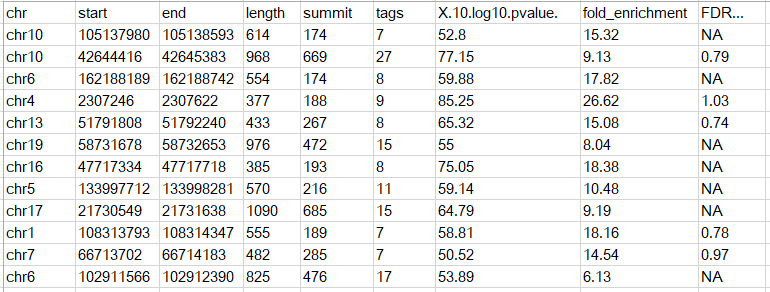
首先我們需要輸入peak文件,支持兩種格式,第一種是BED格式,最少只需要3列內容記錄peak的染色體位置就可以了,示意如下

當然也可以有多余的列,只需要符合BED格式的標準即可;另外一種和MACS的peak calling輸出結果類似,第一行為表頭,示意如下
通過函數readPeaks讀取peak文件,用法如下
peak <- readPeakFile("peak.bed")函數根據文件名稱的后綴來判斷是否為bed格式,建議BED格式的輸入文件后綴統一成.bed, 當然壓縮文件也是支持的,比如.bed.gz;如果不是BED格式的輸入,文件名稱則不能使用BED格式對應的后綴。
下面來詳細看下幾個主要功能的代碼和結果展示
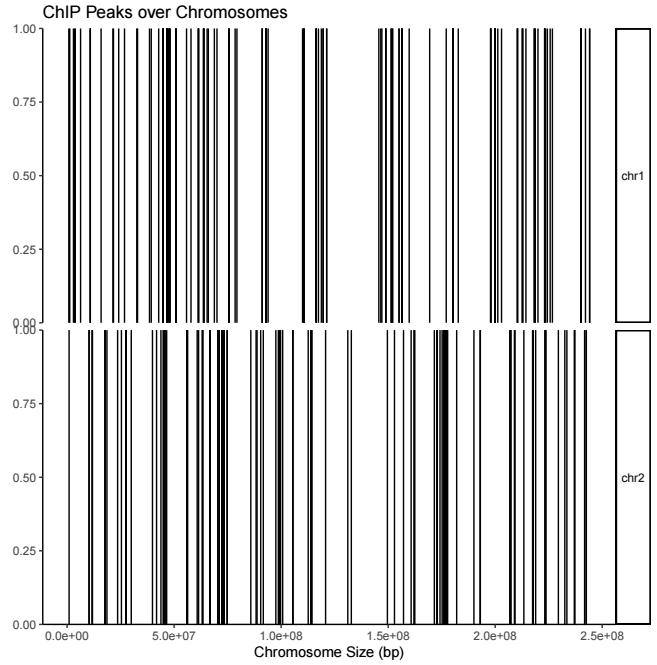
用法如下
covplot(peak, chr = c("chr1", "chr2"))輸出結果示意如下
用法如下
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
# 定義TSS上下游的距離
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrix <- getTagMatrix(peak, windows=promoter)
tagHeatmap(tagMatrix, xlim=c(-3000, 3000), color="red")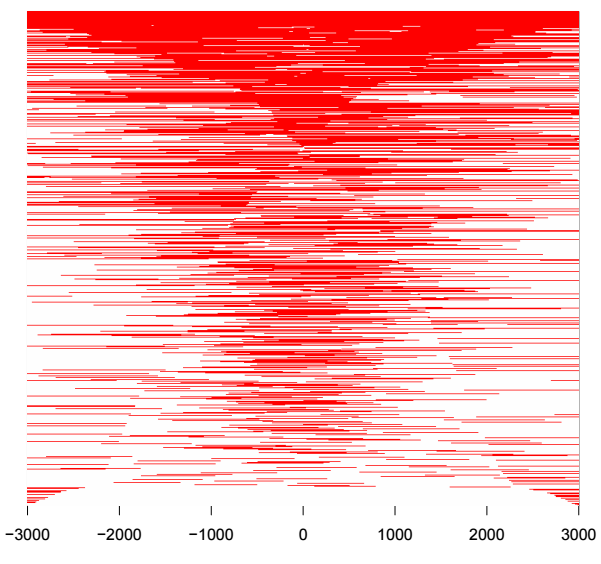
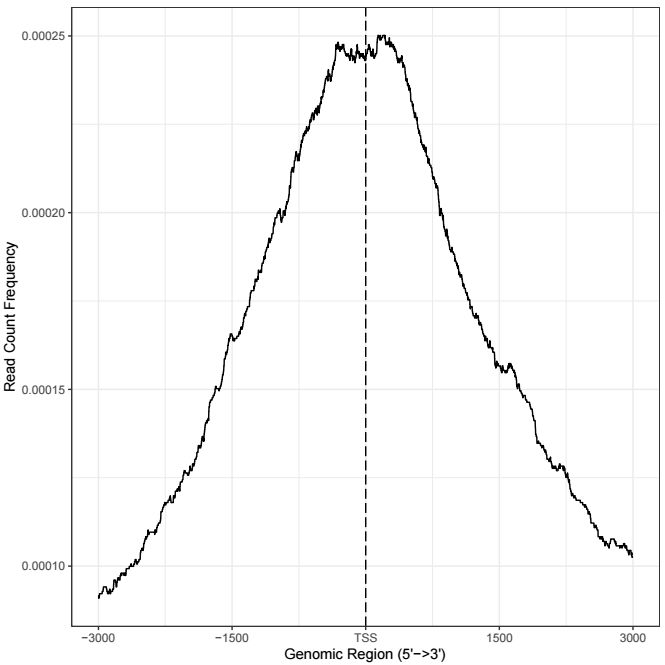
熱圖每一行代表一個基因,展示的是所有基因TSS兩側的分布,除了熱圖外,還可以對所有基因取均值,用折線圖來展示TSS兩側分布情況,用法如下
plotAvgProf(
tagMatrix,
xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency")輸出結果示意如下
用法如下
peakAnno <- annotatePeak(
peak,
tssRegion = c(-3000, 3000),
TxDb = txdb,
annoDb = "org.Hs.eg.db")
write.table(
as.data.frame(peakAnno),
"peak.annotation.tsv",
sep="\t",
row.names = F,
quote = F)注釋文件內容如下
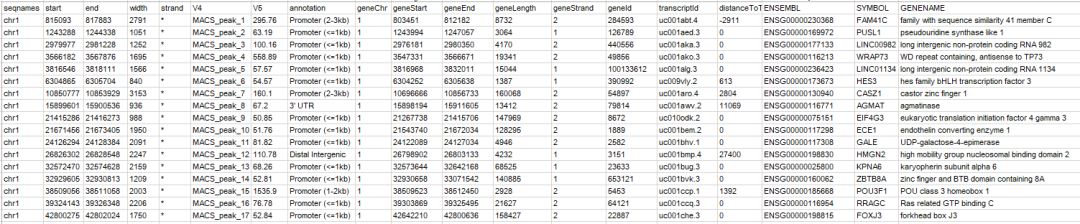
給出了關聯的基因以及對應的基因組區域的類別,根據這個結果,可以提取關聯基因進行下游的功能富集分析,比如提取geneid這一列,用clusterProfiler進行GO/KEGG等功能富集分析。
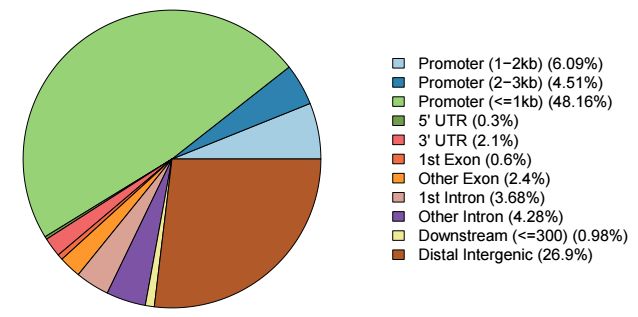
注釋的結果還提供了多種可視化方式,其中餅圖最為常見,用法如下
plotAnnoPie(peakAnno)
輸出結果示意如下
以hg19為例,首先查詢對應的GEO編號信息,用法如下
> hg19 <- getGEOInfo(genome="hg19", simplify=TRUE)
> head(hg19)
series_id gsm organism
111 GSE16256 GSM521889 Homo sapiens
112 GSE16256 GSM521887 Homo sapiens
113 GSE16256 GSM521883 Homo sapiens
114 GSE16256 GSM1010966 Homo sapiens
115 GSE16256 GSM896166 Homo sapiens
116 GSE16256 GSM910577 Homo sapiens由于列數太多,上述結果只展示了部分信息,對于每個bed文件,會列出對應的描述信息,方便篩選感興趣的peak進行下載,可以根據GSM編號進行下載,用法如下
downloadGSMbedFiles("GSM521889", destDir="hg19")也可以根據下載一個基因組對應的所有peak文件,用法如下
downloadGEObedFiles(genome="hg19", destDir="hg19")
peak的overlap分析不僅可以探究生物學重復樣本間的一致性,還可以進一步識別多種蛋白或者轉錄因子在調控網絡中的作用,如果兩個蛋白的chip結果overlap顯著,很可能這兩個蛋白構成了復合體,或者兩種蛋白具有相互作用,這對于探究其調控機制有相當大的幫助。用法如下
enrichPeakOverlap(
queryPeak = peak_setA,
targetPeak = c(peak_setB, peak_setC),
TxDb = txdb,
pAdjustMethod = "BH",
nShuffle = 1000,
chainFile = NULL,
verbose = FALSE)依次將query的peak與target中的每一個peak文件進行overlap分析,計算出一個p值代表兩個peak之間overlap的程度,p值越小,overlap的程度越高。
ChIPseeker除了peak基因注釋的基本功能外,整合了GEO的下載功能與peak的overlap分析,可以方便的將自己的chip_seq數據與GEO的公共數據集進行比較分析。
感謝各位的閱讀,以上就是“如何使用ChIPseeker進行peak注釋”的內容了,經過本文的學習后,相信大家對如何使用ChIPseeker進行peak注釋這一問題有了更深刻的體會,具體使用情況還需要大家實踐驗證。這里是億速云,小編將為大家推送更多相關知識點的文章,歡迎關注!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





