溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
本篇內容主要講解“如何使用HOMER進行peak calling”,感興趣的朋友不妨來看看。本文介紹的方法操作簡單快捷,實用性強。下面就讓小編來帶大家學習“如何使用HOMER進行peak calling”吧!
HOMER是一款進行motif預測的軟件,除此之外,該軟件還集成了許多其他功能,可以識別用于分析chip_seq,RNA_seq,Hi-C等數據。本文主要介紹如何通過HOMER來進行peak calling。
在HOMER中,通過findPeaks這個命令來進行peak calling, 這個命令有以下多種模式,對應不同類型的peak的識別
factor
這種模式用于識DNA和蛋白質結合位點,主要用于識別轉錄因子的結合位點,預測出來的peak的長度是一個固定的數值。
histone
這種模式用于識別發生組蛋白修飾的區域,該模式識別到的peak長度不完全相同,是變化的數值。
super
這種模式用于識別超級增強子。
groseq
這種模式用于分析鏈特異性的GRO_seq數據
tss
這種模式用于分析5’RNA_seq/CAGE/5’GRO_seq, 目的是識別promoter/TSS區域
dnase
這種模式用于分析DNase_seq數據,目的是識別DNase酶超敏位點
mC
這種模式用于識別DNA甲基化區域
對于chip_seq的peak calling而言,常用的模式就是factor, histone和super這3種模式。具體用法如下,分為兩步
比對基因組得到bam文件之后,首先用通過makeTagDirectory這個命令,生成一個文件夾,用法如下
makeTagDirectory out_dir align.bam
輸出目錄文件如下
├── chr1.tags.tsv
├── chr2.tags.tsv
├── chr3.tags.tsv
...
├── chrY.tags.tsv
├── tagAutocorrelation.txt
├── tagCountDistribution.txt
├── tagInfo.txt
└── tagLengthDistribution.txt默認將每條染色體的比對情況有一個tags.tsv文件來存儲,除此之外,還有幾個以tag開頭的文件,包含了一些簡單的統計信息。
tagCountDistribution.txt包含了測序深度的分布信息,第一列為測序深度的值,第二列為對應的reads的比例。根據這個文件的前10行,在R里面可視化如下
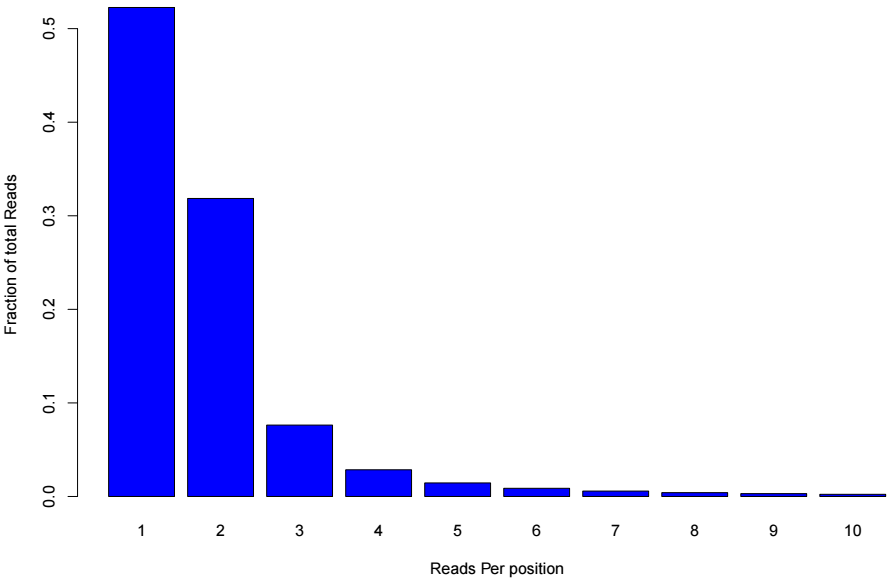
對于chip樣本而言,unique mapping reads的比例越高越好,所以可以看到測序深度為1的比例是最高的。
tagLengthDistribution.txt包含了reads的長度分布信息,第一列為長度,第二列為對應reads的比例, 在R里面可視化如下
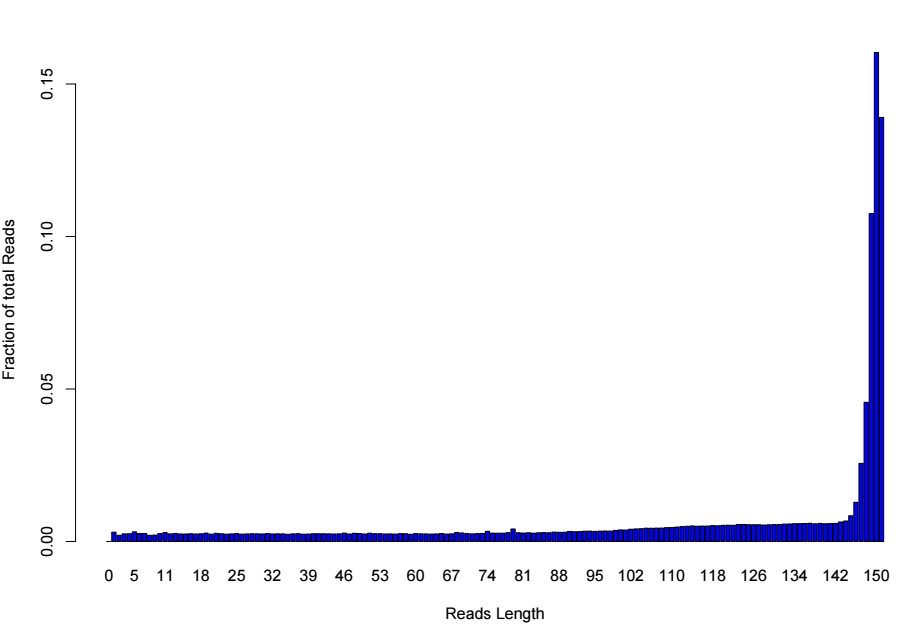
可以對插入片段的長度分布有一個直觀的了解。
tagAutocorrelation.txt用于評估測序數據正負鏈上測序深度分布的相關性,在R里面可視化如下
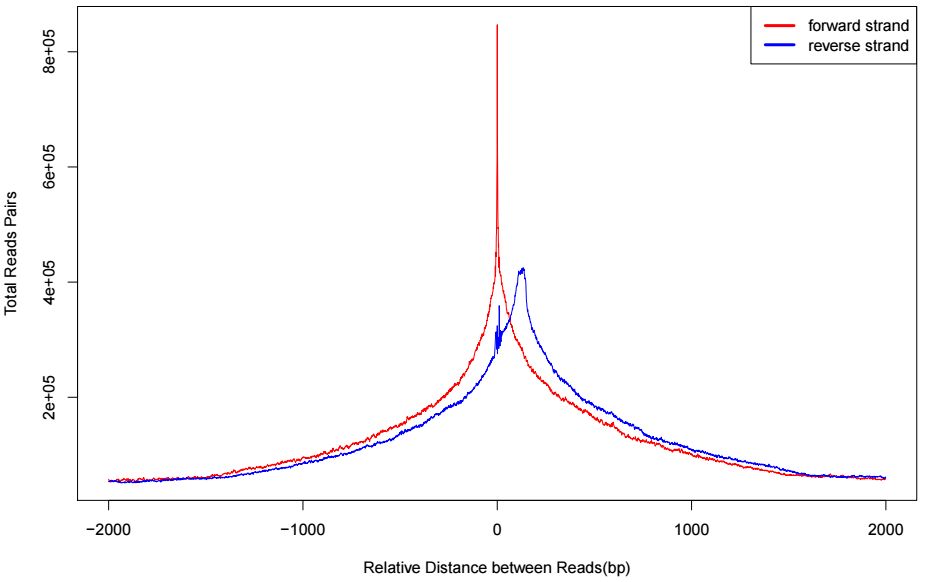
正負連的峰值間距離為插入偏度的長度。
分別對input和IP樣本建立好tagdirectory之后就可以peak calling, 用法如下
findPeaks ip_tagdir/ -i input_tagdir -style histone -o homer.peak.txt
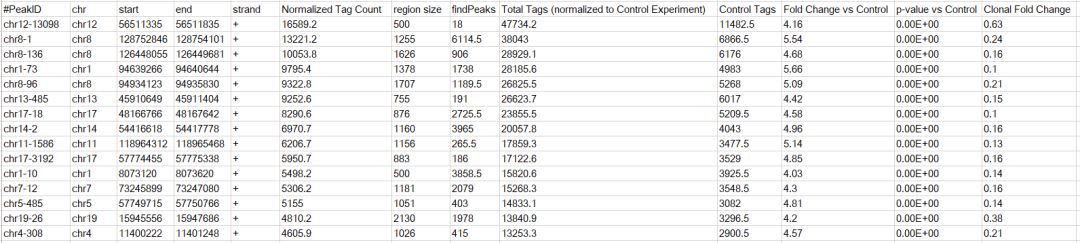
輸出結果和macs2的類似,分成了兩部分,文件頭尾以#開頭的行為注釋行,部分信息如下
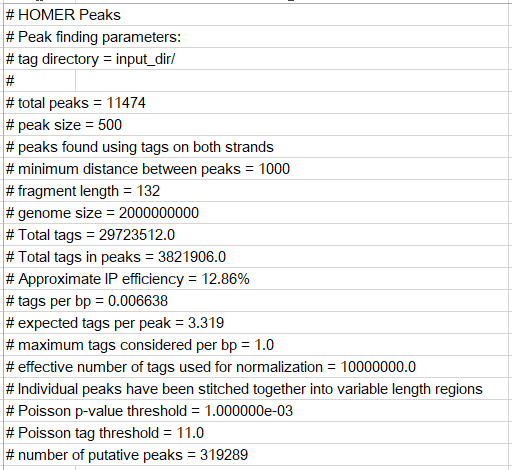
peak對應的行示意如下
 到此,相信大家對“如何使用HOMER進行peak calling”有了更深的了解,不妨來實際操作一番吧!這里是億速云網站,更多相關內容可以進入相關頻道進行查詢,關注我們,繼續學習!
到此,相信大家對“如何使用HOMER進行peak calling”有了更深的了解,不妨來實際操作一番吧!這里是億速云網站,更多相關內容可以進入相關頻道進行查詢,關注我們,繼續學習!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





