溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
如何使用bedtools根據染色體上的起止位置拿到基因symbol,相信很多沒有經驗的人對此束手無策,為此本文總結了問題出現的原因和解決方法,通過這篇文章希望你能解決這個問題。
第一步:將你的染色體位置坐標文件整理成bed格式。
bed格式文件至少包括前3列,分別是:染色體的名字、染色體上的起始位置、染色體上的終止位置。這一步無論用寫字板、excel、R等進行處理都可以,文件的后綴名也不重要,因為強行將文件后綴改為bed時,在后面的Linux系統中進行bedtools處理時也會報錯。所需的bed格式文件參見下圖。
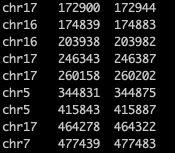
可從gencode中根據自己的需要下載hg38或者hg19版本的人類基因組注釋文件(文章中以hg38為例)。這一步可以進gencode官網(https://www.gencodegenes.org/human/)進行本地下載,然后用filezilla等文件傳輸工具將下載的本地文件傳輸到服務器。也可以直接在服務器的Linux系統中進行ftp下載。
本地下載:

ftp下載:

獲得下載鏈接后,在Linux系統中輸入下面的代碼進行ftp下載:
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_34/gencode.v34.annotation.gtf.gz
在Linux系統中輸入下面的代碼,得到hg38版本的人類蛋白編碼基因的位置坐標:
zcat gencode.v34.annotation.gtf.gz | grep protein_coding |perl -alne '{next unless $F[2] eq "gene" ;/gene_name \"(.*?)\";/; print "$F[0]\t$F[3]\t$F[4]\t$1" }' >protein_coding.hg38.position
先將待處理的坐標bed格式文件鏈接或復制到第三步得到的結果文件所在的目錄下,然后修改這一文件的后綴名為bed,再將這一文件轉化為Tab鍵分隔的后綴名為bed的文件,需輸入下面的代碼(motif1.bed是自己命名的待處理坐標文件):
mv motif1.tsv motif1.bed
perl -p -i -e 's/ /\t/g' motif1.bed
如果在第一步的時候已將待處理的bed格式文件保存為了Tab鍵分隔格式,但是在后面的處理中仍然報錯,不妨再進行一次Tab鍵分隔處理。
首先需要啟動自己安裝了bedtools軟件的conda小環境,然后輸入下面的代碼:
bedtools intersect -a motif1.bed -b ~/dna/exercise/protein_coding.hg38.position -wa -wb
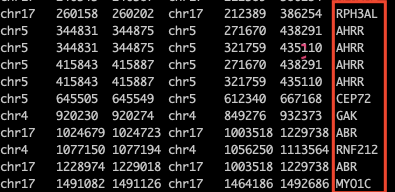
也可以對結果進行匯總,將位于相同染色體坐標的基因symbol寫在一塊,此時只需要加上|后面的代碼即可。| 之前的文件得到的結果有幾列,-c后面的數字就寫幾。如我得到的有7列,-c后面就寫7。
bedtools intersect -a motif1.bed -b ~/dna/exercise/protein_coding.hg38.position -wa -wb | bedtools groupby -i - -g 1-4 -c 7 -o collapse
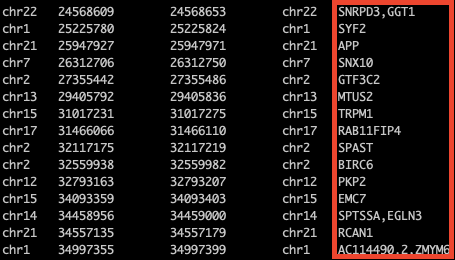
也可以另存結果:
bedtools intersect -a motif1.bed -b ~/dna/exercise/protein_coding.hg38.position -wa -wb | bedtools groupby -i - -g 1-4 -c 7 -o collapse >gene.tsv
新保存的gene.tsv文件就是結果文件了,然后可以拿著結果進行后續處理啦~。
看完上述內容,你們掌握如何使用bedtools根據染色體上的起止位置拿到基因symbol的方法了嗎?如果還想學到更多技能或想了解更多相關內容,歡迎關注億速云行業資訊頻道,感謝各位的閱讀!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





