溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
本篇內容介紹了“tRNAscanSE怎么安裝使用”的有關知識,在實際案例的操作過程中,不少人都會遇到這樣的困境,接下來就讓小編帶領大家學習一下如何處理這些情況吧!希望大家仔細閱讀,能夠學有所成!
安裝方法如下所示:
mkdir tRNAscantar -zxvf trnascan-se-2.0.5.tar.gzcd tRNAscan-SE-2.0./configure --prefix=(path)/tRNAscan #括號中省略了絕對路徑makemake installecho 'export PATH=$PATH:(path)/tRNAscan/bin' >> ~/.bashrcsource ~/.bashrc
wget -c http://eddylab.org/software/infernal/infernal-1.1.2.tar.gzmkdir infernaltar -zxvf infernal-1.1.2.tar.gzcd infernal-1.1.2./configure --prefix=(path)/infernalmakemake installecho 'export PATH=$PATH:(path)/infernal/bin' >> ~/.bashrcsource ~/.bashrc
tRNAscan-SE -B -o tRNA.out -f rRNA.ss -m tRNA.stats genome.fasta-A 適合于古細菌;-B 適合于細菌;-G 適合于古細菌,細菌和真核生物的混合序列;-O 適合于線粒體和葉綠體。不設置以上選項,則默認適合于真核生物;-C 僅使用Cove 進行tRNA 分析。雖然從一定程度上提高了準確性,但是運行速度很慢;-o 結果文件名-f tRNA的二級結構結果文件名-m 統計結果文件名。
對細菌基因組序列進行預測,如下所示:
tRNAscan-SE -B -o twk.tRNA.out -f twk.tRNA.ss -m twk.tRNA.stats new.scaffolds.fasta
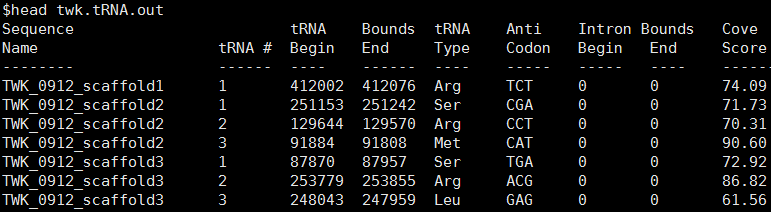
結果文件中out文件為不同Scaffolds上預測的tRNA位置及種類信息:
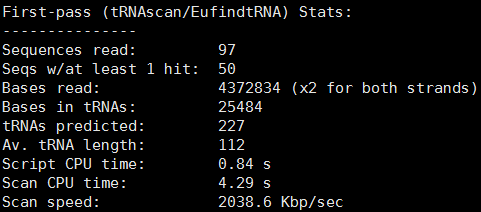
stats文件為預測到的tRNA統計信息,包括預測到的tRNA數、總堿基數等,如下所示:
ss文件為tRNA二級結構信息,如下所示:
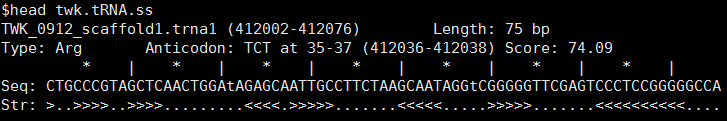
其中大于號或小于號表示互補配對區域,點號表示環形域或非互補配對區域。可以根據out文件與基因組序列提取出tRNA的序列文件與gff文件,如下所示:
perl 10_tRNAscan_parser.pl twk.tRNA.out new.scaffolds.fasta TWK
結果如下所示:

“tRNAscanSE怎么安裝使用”的內容就介紹到這里了,感謝大家的閱讀。如果想了解更多行業相關的知識可以關注億速云網站,小編將為大家輸出更多高質量的實用文章!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





