溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
今天就跟大家聊聊有關SOAPfuse中怎么實現融合基因操作,可能很多人都不太了解,為了讓大家更加了解,小編給大家總結了以下內容,希望大家根據這篇文章可以有所收獲。
和其他軟件類似,SOAPfuse也需要建立物種對應的數據庫,在軟件安裝的source目錄下,提供了建立數據庫的腳本,用法如下
perl SOAPfuse-S00-Generate_SOAPfuse_database.pl \ -wg hg19.fa \ -gtf Homo_sapiens.GRCh47.85.chr.gtf \ -cbd cytoBand.txt.gz \ -gf HGNC_Gene_Family.tsv \ -sd /software/SOAPfuse-v1.27 \ -dd /database/hg19 \ -rft chr.gtp
wg參數代表基因組的fasta文件,gtf參數代表gtf文件,cbd代表從UCSC下載的cytoband文件,gf代表從HGNC下載的基因信息,sd代表軟件的安裝目錄,rft代表gtf文件中的染色體名稱和fasta文件中的染色體名稱的對應關系。
對于需要從數據庫下載的文件,在該腳本的幫助信息中給出了非常詳盡的提示, 這里就不贅述,對于rft文件,內容為\t分隔的兩列,示例如下
1 chr1 2 chr2
第一列代表gtf文件中的染色體編號,第二列代表fasta文件中的染色體編號。
SOAPfuse通過sample.list文件讀取樣本信息,該文件內容如下

\t分隔的4列,第一列代表樣本名稱,第二列代表為lane ID,第三列代表run ID, 第四列代表讀長。之所以每個樣本需要提供lane ID和run ID, 是出于測序時一個樣本會有多條lane的考慮,對于多條lane的數據,因為屬于同一個樣本,所以需要合并起來。
在實際分析時,我們只有每個樣本對應的R1和R2端數據,所以lane ID和run ID自己隨便定義就好了,下面是一個實際的例子,共6例樣本
A1 Lib-A1 Run-A1 150 A2 Lib-A2 Run-A2 150 A3 Lib-A3 Run-A3 150 B1 Lib-B1 Run-B1 150 B2 Lib-B2 Run-B2 150 B3 Lib-B3 Run-B3 150
sample.list中只提供了樣本的名稱等信息,在分析時肯定需要知道每個樣本對應的測序數據的路徑。在SOAPfuse中,通過一個固定的目錄結構來實現,示意如下
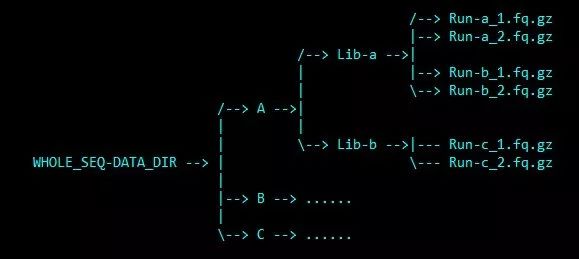
所有的樣本的測序數據位于一個總的目錄下,稱之為WHOLE_SEQ-DATA_DIR,在該目錄下,每個樣本是一個子目錄,名稱必須和sample.list文件中的樣本名一致;在每個樣本的目錄下,是每個lane ID對應的目錄;在lane ID的目錄下,就是樣本的原始數據,以run ID作為前綴。
對于樣本的測序數據,要求是gzip壓縮的格式,支持fasta和fastq兩種格式;文件名稱要求以對應的run ID開頭,雙端數據用_1, _2區分,后綴的話只需要所有樣本統一即可,具體的后綴可以在配置文件中設置。
在軟件安裝的config目錄下,有一個名為config.txt的模板配置文件,我們需要對其進行修改,主要修改以下幾個內容
DB_db_dir = /software/SOAPfuse-v1.27/db/ PG_pg_dir = /software/SOAPfuse-v1.27/source/bin PS_ps_dir = /software/SOAPfuse-v1.27/source PA_all_fq_postfix = fq.gz
DB_db_dir代表第一步建好的數據庫的目錄,后面兩個選項只需要替換成soapfuse實際的安裝目錄就行了,PA_all_fq_postfix代表測序原始數據文件名的后綴,默認是fq.gz。
以上四點內容都準備好之后,就可以進行分析了,代碼如下
perl SOAPfuse-RUN.pl \ -c config.txt \ -fd raw_data \ -l sample.list \ -o out_dir
-c指定配置文件,-fd指定測序數據存放的目錄,-l指定樣本的sample.list文件,-o指定結果的輸出目錄。
看完上述內容,你們對SOAPfuse中怎么實現融合基因操作有進一步的了解嗎?如果還想了解更多知識或者相關內容,請關注億速云行業資訊頻道,感謝大家的支持。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





