溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
htseq-count中怎么實現定量分析操作,很多新手對此不是很清楚,為了幫助大家解決這個難題,下面小編將為大家詳細講解,有這方面需求的人可以來學習下,希望你能有所收獲。
和featurecounts一樣,htseq-count也是一款進行raw count定量的軟件。該軟件采用python語言進行開發,集成在HTseq這個包中。
對于python的包,通過pip可以方便的進行安裝,代碼如下
pip install HTSeq
HTSeq提供了許多處理NGS數據的功能,htseq-count只是其中進行定量分析的一個模塊。
htseq-count的設計思想和featurecounts非常類似,也包含了feature和meta-features兩個概念。對于轉錄組數據而言,feature指的是exon, 而meta-feature可以是gene, 也可以是transcript。
進行定量分析需要以下兩個文件
比對的BAM/SAM文件
基因組的GTF文件
對于雙端數據,要求輸入sort之后的BAM文件。
由于序列讀長的限制和基因組的同源性,一條reads可能比對到多個基因上,而且基因之間也存在overlap, 在對這些特殊情況進行處理時,htseq-count內置了以下3種模式
union
intersection-strict
intersection-nonempty
通過--mode參數指定某種模式,默認值為union。這3種模式在判斷一條reads是否屬于某個feature時,有不同的判別標準,示意圖如下
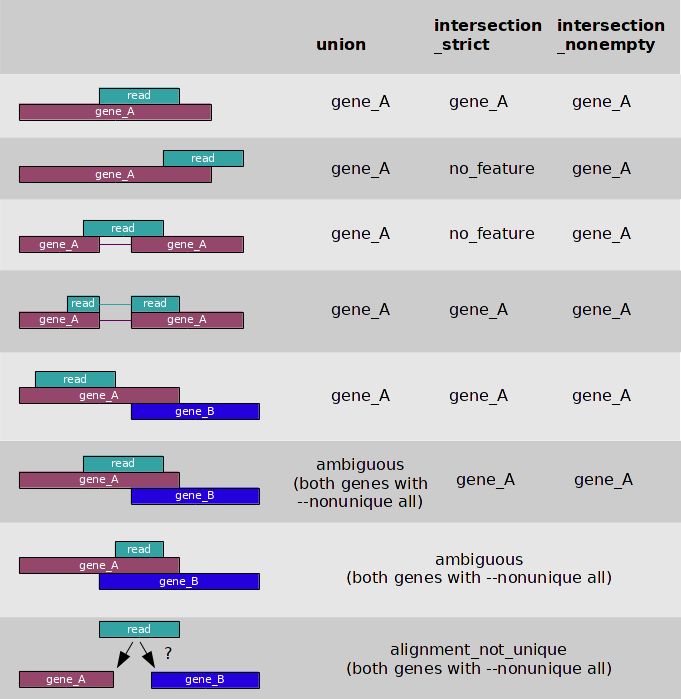
在BAM文件,包含了比對上的reads和沒有比對上的reads, 只有比對上的reads 會用來計數,htseq-count默認會根據mapping的質量值對BAM文件進行過濾,默認值為10, 意味著只有mapping quality > 10的reads才會用來計數,當然可以通過-a參數來修改這個閾值。
能夠明確reads屬于一個featurer時,比如示意圖種的第一種情況,reads完全是gene_A的一個片段,該feature的計數就加1;能明確reads不屬于一個feature時,稱之為no_feature, 比如示意圖種的第二種情況,reads的一部分比對上了gene_A, 在intersection_strict模式下,判定該reads不屬于gene_A, 就是no_feature。
當不明確一條reads是否屬于某個feature時,通常是由于reads在兩個gene的overlap區,比如示意圖中的第六和第七種情況,這樣的reads被標記為ambigous。
當一條reads比對上了兩個feature時,會被標記為alignment_not_unique。
在統計屬于某個基因的reads數時,需要重點關注對 ambiguous 和 alignment_not_unique 的reads的處理, 通過--nonunique參數來指定,取值有以下兩種
none
all
默認值為none時,這兩種reads被忽略掉,不進行任何的計數;取值為all時,對應的所有feature的計數都會加1。
除了--mode和--nonunique兩個參數外,還需要關注--stranded參數,這個參數指定文庫的類型,默認值為yes, 代表文庫為鏈特異性文庫,no代表為非鏈特異性文庫。對于非鏈特異性文庫文庫,在判斷一條reads是否屬于一個基因時,只需要關注比對位置,而鏈特異性文庫還需要關注比對的正負鏈和基因的正負鏈是否一致,只有一致時,才會計數。
理解了以上3個參數,就能夠正確的使用htseq-count了。對于非鏈特異性的數據,常規用法如下
htseq-count \ -f bam \ -r name \ -s no \ -a 10 \ -t exon \ -i gene_id \ -m union \ --nonunique=none \ -o htseq.count \ align.sorted.bam \ hg19.gtf
在運行速度上,featurecounts比htseq-count快很多倍,而且feature-count不僅支持基因/轉錄本的定量,也支持exon等單個feature的定量。所以更加推薦使用featurecounts來定量。
看完上述內容是否對您有幫助呢?如果還想對相關知識有進一步的了解或閱讀更多相關文章,請關注億速云行業資訊頻道,感謝您對億速云的支持。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





